2024-02-03
Here is a recent paper with Melissa Jungnickel and John Hickey:
Johnsson, M., Hickey, J.M. & Jungnickel, M.K. Building in vitro tools for livestock genomics: chromosomal variation within the PK15 cell line. BMC Genomics 25, 49 (2024).
The context is this: We and many others have been doing these large genome-wide association studies of farm animal populations (like Gozalo-Marcilla et al. 2021, Desire et al. 2023), and sequencing their genomes to find lots of variants (like in Ros-Freixedes et al. 2022). To connect sequence variants to associations, we would like to do high-throughput molecular assays. They need cell lines. So we looked to one of the classic, arguably the classic cell line for pigs: PK15.
We sequenced a couple of isolates – one fresh from the American Type Culture Collection and one that had been used in the lab for a long time – and like others have found (for example in farm animal cell lines: de Vos et al. 2023), they had several aneuploidies and large structural variants in their genomes. The aneuploidies are partially different, and the lab sample has more. We also looked for anueploidy in publicly available RNA sequencing data. Those results should be interpreted more cautiously because of the added uncertainty of variation in expression level, but they look like they have aneuploidies as well. For comparison, we sequenced a recently isolated fibroblast line, and its genome looked fine.
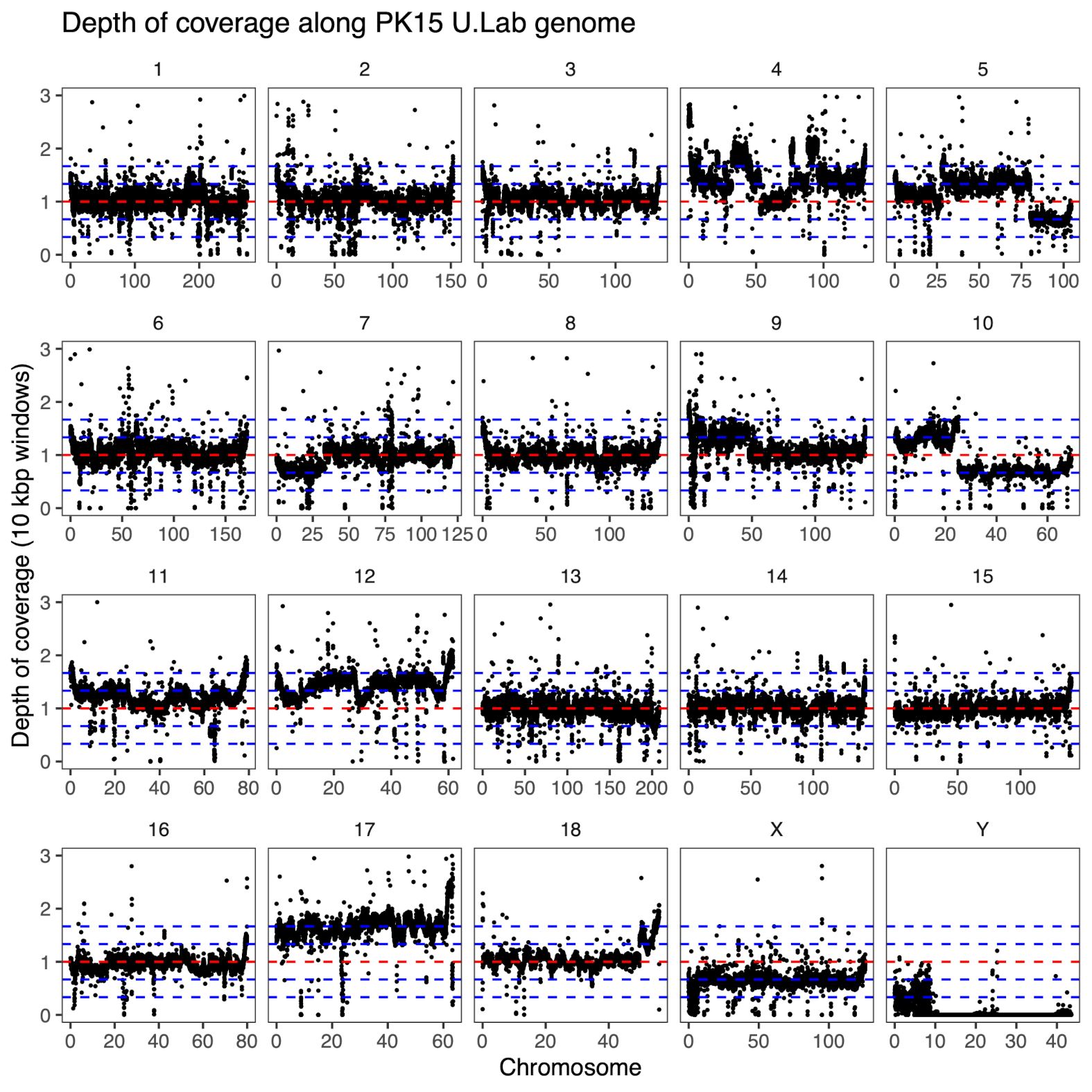
We looked at the relative depth of coverage and within-sample allele frequencies. The relative coverage measures how many sequence reads that align to a segment compared to the median of the whole genome (figure below); in a tetrasomic chromosome you would expect 3/2 the depth of coverage of a regular disomic chromosome. The within-sample allele frequency is based on single nucleotide variants detected as heterozygous by the variant caller. For a variant on a tetrasomic chromosome you would expect allele frequencies close to 1/3 and 2/3, compared to frequencies close to 1/2 on a regular disomic chromosome – because there is one more chromosome copy, that will carry either of the alleles.
There are some puzzling chromosomes. For example, in the university lab isolate, the presence of only one allele for most variants on the X chromosome makes it look like there is only one copy, but the depth of coverage is not 1/2 of the average, but about 2/3. Chromosome 17, on the other hand, has very high depth of coverage and a within-sample allele frequency relationship that looks more like it’s pentasomic. In fact, most chromosomes in the university lab sample have allele frequency modes away from 1/2, suggesting additional copies of most of the genome.
We talk only a little about it in the supplementary materials, but I spent some time simulating different scenarios to think about how these patterns can come about1. There are tools to infer structural variation in a more systematic way, but we can’t expect them to do well, because with the combination of aneuploidy and clonal heterogeneity, there are multiple ways to create the same pattern. It might have been interesting to do some actual measurements to look at the karyotypes and variation within the cell populations. But it’s also a little beside the point to investigate the particular karyotype of a single isolate, when it seems to vary.
Another thing we didn’t mention in the paper but I think is interesting: at the population sizes that these cells are kept, even during passaging, new variants are unlikely to get fixed unless they are beneficial or the mutation rate is huge and asymmetrical. This suggests that the aneuploidies are adaptations to growth in culture.
The story of the paper itself is one of almost not making it into print. All three authors have moved on to other things, and after BMC Genomics sat on the paper for a long time, if the requests for revisions had been more demanding, we would not have been able to do them. But the paper made it, so here it is!
I could blog about it on a rainy day (i.e., probably never).↩︎